
|
|
      |
Hot keys | Text commands | Amino Acids property scales | Custom residue coloring Here is the expanded list of features provided by our custom VMD startup file. If the startup file was read by VMD, the brief version of this description can be launched in Tkconsole by entering 'h' command. In addition to keys and commands the startup file presets the default directory to C:/Temp, relocates the windows and menus so that they fit to 1024x768 screen, hides x-y-z axis arrows and modifies coloring by the residue name so that it uses colors in a manner similar to the residue type, but with larger number of colors. Hot Keys (for OpenGL Window):Mouse modeR enter rotate mode; stop rotation T enter translate mode S enter scaling mode C assign rotation center 0 query item; show labels menu 1 pick atom 2 pick bond (2 atoms) 3 pick angle (3 atoms) 4 pick dihedral (4 atoms) 5 move atom 6 move residue 7 move fragment 8 move molecule 9 move highlighted rep ViewQ view from positive direction of x axis (|||screenshot) W view from positive direction of y axis (|||screenshot) E view from positive direction of z axis (|||screenshot) F flip view 180º (view from the back of the current view) X spin about x axis Y spin about y axis Z spin about z axis J rotate 2º about x K rotate -2º about x L rotate 2º about y H rotate -2º about y RepresentationsN apply preselected graphical representation (new ribbons colored by index) (|||screenshot) I apply preselected graphical representation (trace colored by index) (|||screenshot) V set white background and 'exp2' depth cue (|||screenshot) B set black background without depthcue P switch depthcue on and off U make the selections of the top molecule to auto update each frame A apply representations from the top molecule to all other molecules. Based on save_state script by John Stone. Additional graphicsO (o) draw coordinate cylinders in origin (red x, green y, blue z) (|||screenshot) G draw coordinate greed (red x, green y, blue z). One tick 1Å, small square 5Å, big square 10Å. The whole greed is +- 100 A. It looks neat when used with view from x, y, or z directions (buttons Q,W,E) (|||screenshot) D remove all the graphics added Menus[ show main menu (|||screenshot) ] show files menu, and set the current folder as of the top molecule file (|||screenshot) ' show graphics (Graphical Representations) menu (|||screenshot) \ show sequence menu (|||screenshot) ; show tkcon Tcl console (Works after the first use of Extensions -> tkcon) (|||screenshot) Animation+ move to next frame - move to previous frame . > play animation forward , < play animation reverse / ? stop animation ModificationsM move geometry center of the molecule to the origin ` ~ orient top molecule (not more than 50,000 atoms) by principal axes (requires Orient script written by Paul Grayson and requires linear algebra package by Hume Integration Software) (|||screenshot) Text Commands (for the console):h Show this list of Hot keys and Text commands
a Loads coordinates from the filelist and adds them as new frames to the top molecule
Syntax: a {file1.coor file2.pdb file3.dcd}; a {c:/temp} {file1.dcd} 10
Filenames can be separated by new lines. First argument can be path to files, it can be
omitted or empty. The third argument can be step for trajectory reading or can be omitted.
Filename can include full path.
n Loads coordinates from the filelist and adds them as new files.
Syntax: n {file1.coor file2.pdb file3.dcd}; n {c:/temp} {file1.dcd} 10
Filenames can be separated by new lines. First argument can be path to files, it can be
omitted or empty. The third argument can be step for trajectory reading or can be omitted.
t sets current working folder to 'C:/Temp'
c sets current working folder to the path to the first loaded file of the top molecule
pdb Write current frame of the top file to a pdb file <old_name>_.pdb
fx Rotate (flip) protein 180 degrees around x axis (changes coordinates)
fy Rotate (flip) protein 180 degrees around y axis (changes coordinates)
fz Rotate (flip) protein 180 degrees around z axis (changes coordinates)
colstart Starts collaboration session between two VMD machines
Based on Justin Gullingsrud's vmdcollab script
colstop Stops collaboration session between two VMD machines
Based on Justin Gullingsrud's vmdcollab script
betascale Sets beta value of protein residues to the value found in the
selected hydrophobicity scale. (|||screenshot)
Usage: 'betascale' - prints the list of
scales; 'betascale <scale_name>' - assigns values from the scale ;
'betascale <scale_name> scale' - assigns values from the scale and
prints scale values on the console screen.
Most of the scales are taken from the ExPASy ProtScale page.
bs Same as 'betascale'
mutant Creates mutant of the protein from the current structure using PSFGEN. (|||screenshot)
Usage: 'mutant <resnum> <mutant_restype>' E.g.: 'mutant 109 THR 115 VAL'
morph Adds frames with linear interpolation between the existing frames of the top
molecule. Usage: 'morph <frames_increase_factor> <frames_insertion_frequency_type>'
or 'morph <start:frames_increase_factor:end> <frames_insertion_frequency_type>'
E.g. 'morph 10' 'morph 2 linear' 'morph 3 cycle' 'morph 100 sin2' 'morph 3:10:4 linear'
based on the "morph 1.0" by Andrew Dalke (dalke@ks.uiuc.edu), and modified
to handle the trajectory with any number of frames
cell Sets the cell size for periodic images.
Usage: cell <{Specified a, b, c, alpha, beta, gamma}|{line from xsc file}|{cell
parameters from NAMD configuration}|{xsc filename}>
E.g. 'cell {100 110 120 90 90 60}' 'cell {sim-mscs-007.xsc}'
symm calculate symmetric structure closest to the starting structure of homooligomer,
or spread conformation of one monomer onto the whole oligomer. (|||screenshot)
Usage: symm <|"selection_string"> <|monomer_index|symmetrization_mode>.
monomer_index: integer 0 to (number_of_monomers - 1)
symmetrization_mode: 'avg' - average, 'max' - the most different monomer,
'min' - monomer closest to the average. E.g. 'symm' 'symm "resid 23"'
'symm min' 'symm max' 'symm avg' 'symm "resid 23" 0'
'symm' is equivalent to 'symm "protein" max'
radii sets atomic VDW radii according to the values read from the predefined values,
or from file with atom types and radii, or from CHARMM parameters file. (|||screenshot)
Syntax: radii {} ==> Displays brief help and sets predefined CHARMM radii
radii v ==> restores default VMD radii
radii {c:/path/param_charmm.inp} ==> reads radii from CHARMM parameters file
radii {c:/path/raddi_file.txt} r ==> reads radii from 'type TAB radius' file
ss starts sscache script recalculating secondary structure for every frame and keeping it
in the cache for fast use. Based on the sscache script by Andrew Dalke
(dalke@ks.uiuc.edu)
Usage: ss <molid|top|>. E.g. 'ss', 'ss top', 'ss 2'
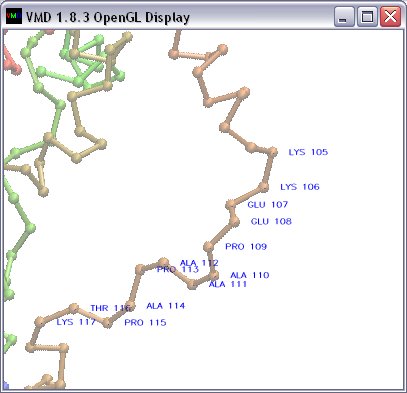
lbl Labels each atom in the 'selectionText' with information 'labelInfo', with
arbitrary prefix 'labelPrefix', color 'color' and font size 'size' (default 1). (|||screenshot)
Syntax: 'lbl <|selectionText> <|labelInfo> <|labelPrefix> <|color> <|size>'
E.g. 'lbl {resid 1 to 10 and name CA}',
'lbl {name CA} {resname resid} { } blue 0.5'
58 Amino Acid Property Scales (for 'bs' command):Most of the scales are taken from the ExPASy ProtScale page. Numerical values fro the scales can be either checked out directly in the vmd.rc file (search for the procedure 'betascale'), or can be seen in Tkconsole if you enter 'betascale <scale_name> scale' or 'bs <scale_name> scale' command. AA_Composition ---Overall amino acid composition (%). (McCaldon P., Argos P.) AA_SwissProt ---Amino acid composition (%) in the Swiss-Prot Protein Sequence data bank. (Bairoch A.) AccessibleResidues ---Molar fraction (%) of 3220 accessible residues. (Janin J.) AlphaHelix_Fasman ---Amino acid scale: Conformational parameter for alpha helix (computed from 29 proteins). ( Chou P.Y., Fasman G.D.) AlphaHelix_Levitt ---Normalized frequency for alpha helix. (Levitt M.) AlphaHelix_Roux ---Conformational parameter for alpha helix. (Deleage G., Roux B.) AntiparallelBetaStrand ---Conformational preference for antiparallel beta strand. (Lifson S., Sander C.) AverageBuried ---Average area buried on transfer from standard state to folded protein. (Rose G.D., Geselowitz A.R., Lesser G.J., Lee R.H., Zehfus M.H.) AverageFlexibility ---Average flexibility index. (Bhaskaran R., Ponnuswamy P.K.) BetaSheet_Fasman ---Conformational parameter for beta-sheet (computed from 29 proteins). (Chou P.Y., Fasman G.D.) BetaSheet_Levitt ---Normalized frequency for beta-sheet. (Levitt M.) BetaSheet_Roux ---Conformational parameter for beta-sheet. (Deleage G., Roux B.) BetaTurn_Fasman ---Conformational parameter for beta-turn (computed from 29 proteins). (Chou P.Y., Fasman G.D.) BetaTurn_Levitt ---Normalized frequency for beta-turn. (Levitt M.) BetaTurn_Roux ---Conformational parameter for beta-turn. (Deleage G., Roux B.) Bulkiness ---Bulkiness. (Zimmerman J.M., Eliezer N., Simha R.) BuriedResidues ---Molar fraction (%) of 2001 buried residues. (Janin J.) Coil_Roux ---Conformational parameter for coil. (Deleage G., Roux B.) Hphob_Argos ---Membrane buried helix parameter. (Rao M.J.K., Argos P.) Hphob_Black ---Amino acid scale: Hydrophobicity of physiological L-alpha amino acids ( Black S.D., Mould D.R.) Hphob_Breese ---Hydrophobicity (free energy of transfer to surface in kcal/mole). (Bull H.B., Breese K.) Hphob_Chothia ---Proportion of residues 95% buried (in 12 proteins). (Chothia C.) Hphob_Doolittle ---Hydropathicity. (Kyte J., Doolittle R.F.) Hphob_Eisenberg ---Normalized consensus hydrophobicity scale. (Eisenberg D., Schwarz E., Komarony M., Wall R.) Hphob_Fauchere ---Hydrophobicity scale (pi-r). (Fauchere J.-L., Pliska V.E.) Hphob_Guy ---Hydrophobicity scale based on free energy of transfer (kcal/mole). (Guy H.R.) Hphob_Janin ---Free energy of transfer from inside to outside of a globular protein. (Janin J.) Hphob_Leo ---Amino acid scale: Hydrophobicity (delta G1/2 cal) ( Abraham D.J., Leo A.J.) Hphob_Manavalan ---Average surrounding hydrophobicity. (Manavalan P., Ponnuswamy P.K.) Hphob_Miyazawa ---Hydrophobicity scale (contact energy derived from 3D data). (Miyazawa S., Jernigen R.L.) Hphob_mobility ---Mobilities of amino acids on chromatography paper (RF). (Aboderin A.A.) Hphob_Parker ---Hydrophilicity scale derived from HPLC peptide retention times. (Parker J.M.R., Guo D., Hodges R.S.) Hphob_pH3.4 ---Hydrophobicity indices at ph 3.4 determined by HPLC. (Cowan R., Whittaker R.G.) Hphob_pH7.5 ---Hydrophobicity indices at ph 7.5 determined by HPLC. (Cowan R., Whittaker R.G.) Hphob_Rose ---Mean fractional area loss (f) [average area buried/standard state area]. (Rose G.D., Geselowitz A.R., Lesser G.J., Lee R.H., Zehfus M.H.) Hphob_Roseman ---Hydrophobicity scale (pi-r). (Roseman M.A.) Hphob_Sweet ---Optimized matching hydrophobicity (OMH). (Sweet R.M., Eisenberg D.) Hphob_Welling ---Antigenicity value X 10. (Welling G.W., Weijer W.J., Van der Zee R., Welling-Wester S.) Hphob_Wilson ---Hydrophobic constants derived from HPLC peptide retention times.(Wilson K.J., Honegger A., Stotzel R.P., Hughes G.J.) Hphob_Wolfenden ---Hydration potential (kcal/mole) at 25øC. (Wolfenden R.V., Andersson L., Cullis P.M., Southgate C.C.F.) Hphob_Woods ---Hydrophilicity. (Hopp T.P., Woods K.R.) HPLC2.1 ---Retention coefficient in HPLC, pH 2.1. (Meek J.L.) HPLC7.4 ---Retention coefficient in HPLC, pH 7.4. (Meek J.L.) HPLCHFBA ---Retention coefficient in HFBA. (Browne C.A., Bennett H.P.J., Solomon S.) HPLCTFA ---Retention coefficient in TFA. (Browne C.A., Bennett H.P.J., Solomon S.) MolecularWeight ---Molecular weight of each amino acid. NumberCodons ---Number of codon(s) coding for each amino acid in universal genetic code. ParallelBetaStrand ---Amino acid scale: Conformational preference for parallel beta strand. ( Lifson S., Sander C.) Polarity_Grantham ---Polarity (p). (Grantham R.) Polarity_Zimmerman ---Polarity. (Zimmerman J.M., Eliezer N., Simha R.) RatioSide ---Atomic weight ratio of hetero elements in end group to C in side chain. (Grantham R.) RecognitionFactors ---Recognition factors. (Fraga S.) Refractivity ---Refractivity. (Jones. D.D.) RelativeMutability ---Relative mutability of amino acids (Ala=100). (Dayhoff M.O., Schwartz R.M., Orcutt B.C.) TotalBetaStrand ---Conformational preference for total beta strand (antiparallel+parallel). (Lifson S., Sander C.) Hphob_Privalov_dCp ---deltaCp hydration, J/(K*mol*A^2), (Privalov, P. L. & Khechinashvili, N. N.) Hphob_Privalov_dH ---deltaH hydration, J/(mol*A^2), 25 oC (Privalov, P. L. & Khechinashvili, N. N.) Hphob_Privalov_dS ---deltaS hydration, J/(K*mol*A^2), 25 oC (Privalov, P. L. & Khechinashvili, N. N.) Hphob_Privalov_dG ---deltaG hydration, J/(mol*A^2), 25 oC (Privalov, P. L. & Khechinashvili, N. N.) Custom coloring by residue nameALA gray ARG blue ASN green ASP red CYS yellow GLY black GLU red GLN lime HIS mauve ILE white LEU white LYS blue MET orange PHE pink PRO cyan SER tan THR ochre TRP purple TYR purple VAL silver TIP iceblue TIP3 iceblue TP3E iceblue WAT iceblue SOL iceblue H2O iceblue ZN blue NA blue CL red CLA red POT red SOD red |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}