Lab
12.
SDS-polyacrylamide gel eletrophoresis and
Western blot
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) is one of the methods used to resolve different proteins in a complex mixture. Western blot analysis is used to identify a particular protein in a complex mixture. These techniques are used extensively in research laboratories to detect and quantify a particular protein. These techniques are also useful in clinical diagnosis for the detection of specific antibodies in serum or proteins of disease agents in clinical specimens. For example, HIV in a clinical sample can be detected by the presence of viral structural proteins using electrophoresis and Western blot analysis.There are 3 stages in immunodetection of proteins by Western blotting:
1. SDS-PAGE
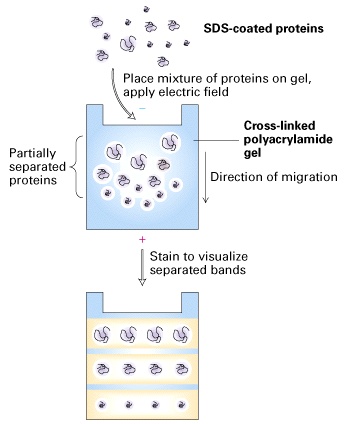
In this stage, each component of a protein mixture is resolved by SDS-PAGE as a separate band. Highly cross-linked polyacrylamide forms a gel matrix through which proteins migrate. The mobility of proteins in a gel is dependent on their sizes and the concentration of acrylamide. The gel acts as a molecular sieve. Small proteins move through the gel faster than big proteins. The higher the acrylamide concentration is, the smaller the pores of the gel are, and the slower proteins move. Therefore, gels with high concentrations of acrylamide are used to resolve small sizes of proteins, and gels with low concentrations of acrylamide are used to resolve big sizes of proteins.Before electrophoresis, samples are treated with a solution containing b-mercaptoethanol and sodium dodecyl sulfate (SDS). b-mercaptoethanol is a reducing agent that cleaves disulfide bonds. SDS is an anionic detergent that denatures proteins and coats proteins with negative charges, which overwhelm the protein’s intrinsic charge. Thus, treated proteins assume a rod-like shape and all carry the same net negative charge. When treated proteins are electrophoresed through a SDS-PAGE gel, proteins migrate toward the positive electrode. The proteins can now be separated by their molecular weights.
The SDS-PAGE gel that you will prepare for this lab exercise has two layers. The top layer is stacking gel, and the bottom layer is running gel. Stacking gel has a relatively low concentration of acrylamide and a lower pH, and running gel has a relatively high concentration of acrylamide and a high pH. Because of the low concentration of acrylamide, proteins of different sizes move at the same pace in the stacking gel. When proteins hit the running gel that has a high concentration of acrylamide, proteins are stacked at the border between the stacking and running gel before moving forward, which makes resolved protein bands sharp and provides a better resolution.
SDS-PAGE electrophoresis is a more powerful procedure than chromatography because it can be used to separate all types of proteins, including membrane proteins, the cytoskeleteton proteins and protein aggregates that normally are insoluble in water. In addition, SDS-PAGE electrophoresis can be used to separate proteins in a small quantity.
2. Transfer
Before Western blot analysis, proteins resolved by electrophoresis are transferred from the gel to nitrocellulose membrane electrophoretically. During the transfer, the gel is at the negative electrode side and the nitrocellulose membrane at the positive electrode side. Proteins that are coated with negatively charged SDS will move from the negative side, the gel, to the positive side, the nitrocellulose.The advantages of transferring the protein to a membrane include: 1) ease of handling relative to the gel, 2) the ability to probe the membrane, 3) the membrane can be stored, and potentially stripped, and reprobed, 4) protein quantification is facilitated.
3. Western blot
Western blot, also called immunoblot, uses antibody to identify a specific protein of interest. Similar to ELISA, the nitrocellulose membrane that contains the resolved proteins will be first incubated with an unrelated protein to block nonspecific protein binding sites on the membrane, and then blotted with a primary antibody that is specific for the protein of interest. The unbound primary antibody will be washed away. The solution used for washing contains a detergent and a high concentration of salt that eliminate nonspecific binding of the antibody to the nitrocellulose through hydrophobic or ionic interaction. Next is the addition of a secondary antibody that is specific for the primary antibody and conjugated to horse reddish peroxidase (HRP). After washing, a substrate of HRP will be added. HRP catalyzes a reaction that transforms the colorless substrate into a colored product, which visualizes the protein band.Gel electrophoresis/Western blot is one of the powerful methods for detecting a particular protein in a complex mixture. This technique combines the superior resolving power of gel electrophoresis, the specificity of antibodies, and the sensitivity of enzyme assays. Compared to gel electrophoresis/Western blot, ELISA is better for the quantification rather than identification of a protein of interest.
From Molecular Cell Biology by Harvey Lodish et al.Procedure
March 29 Casting discontinuous SDS-PAGE gel
1. Clean glass plates with wet kimwipes. Please be careful when you handle the glass plates.
2. Assemble the glass plate sandwiches as demonstrated by the TAs.
3. Place a comb completely into the assembled gel sandwich. With a marker pen, place a mark on the glass plate 1 cm below the teeth of the comb. This will be the level to which the separating gel is poured. Remove the comb.
4. Prepare the separating gel monomer solution by combining all reagents except ammonium persulfate (APS) and TEMED.
Unpolymerized acrylamide is a neurotoxin, so please wear gloves when you handle it!!!
Separating gel 10%
Components Volume Acrylamide/BIS (40% stock)* 2.76 ml 1.5 M Tris-HCl, 0.5% SDS, pH 8.8 3.08 ml 0.5 M Tris-HCl, 0.5% SDS, pH 6.8 - dH2O 5.4 ml 3% ammonium persulfate 0.2 ml TEMED 7 ml *Acrylamide/BIS = 37.5:1 5. Add APS first and then TEMED to the monomer solution. Mix and pour the solution to the mark, using a 5 ml pipette. Please pour the solution down the side of one of the spacers. Pour the solution smoothly to prevent it from mixing with air and forming bubbles.
6. Immediately overlay the monomer solution with water using a p200 pipetteman. It must be carried out slowly, with a steady, even rate of delivery to prevent mixing. Add 100 ml of distilled water from each side of the gel.
7. Allow the gel to polymerize for 45 minutes, and then rinse the top of the gel twice with distilled water.
8. Prepare the stacking gel monomer solution. Combine all reagents except APS and TEMED.
Stacking gel 4.5%
Components Volume Acrylamide/BIS (40% stock)* 0.56 ml 1.5 M Tris-HCl, 0.5% SDS, pH 8.8 - 0.5 M Tris-HCl, 0.5% SDS, pH 6.8 1.25 ml dH2O 3.15 ml 3% ammonium persulfate 0.05 ml TEMED 5 ml *Acrylamide/BIS = 37.5:1 9. Drain the water on the top of the separating gel before pouring the stacking gel.
10. Place a comb in the gel sandwich, and tilt it a little so the comb can easily get in and no air is trapped under the comb teeth when the monomer solution is poured.
11. Add APS and TEMED to the monomer solution, mix, and pour the solution down the spacer nearest the upturned side of the comb. Pour all the way to the top. Then properly align the comb in the sandwich.
12. After the gel polymerizes, your TA will wrap your gel with wet paper towel and Saran wrap. The gels will be stored at 4oC for Wednesday.
March 31 Electrophoresis and Transfer
Part 1. Electrophoresis
Solutions:
Sample buffer (SB):
0.5 M Tris-HCl, pH 6.8 1.0 ml
SDS 0.2 g
2-mercaptoethanol 0.5 ml
glycerol 1.0 ml
0.05% bromophenol blue 0.5 ml
dH2O 6.8 mlGel running buffer
0.5 M Tris base, pH 8.0
0.5 M glycine
1% SDSSample preparation:
1. Add 20 ml of sample buffer to each of your samples from the last lab exercise.2. In an eppendorf tube, combine 10 ml of 10 mg/ml BSA with 30 ml of sample buffer. In another eppendorf combine 5 ml of pre-stain molecular weight markers with 10 ml of the sample buffer.
3. Boil all eppendorfs for 5 minutes and cool.
4. Spin all your samples in a microcentrifuge at full speed for 2 min.
5. Wash the wells with the gel-running buffer twice and fill the wells with the gel running buffer.
Loading gel:
6. Load the gel with loading tips.
Well 1 2 3 4 5 6 7 8 9 10 Sample SB MW IP+ IP- IP+ IP- BSA BSA BSA SB ml 15 15 15 15 15 15 15 10 5 15 Running gel:
7. Run the gel at 30 mA for one gel and 60 mA for two gels.Part 2. Transfer
Solution:
Transfer buffer
Tris base 3.03 g
Glycine 144 g
Methonal 200 ml
dH2O up to 1 L1. Remove your gel as demonstrated by the TAs, and put the gel in a pan of transfer buffer.
2. Wearing gloves, cut a piece of nitrocellulose to about the same size of the gel. In a corner of the nitrocellulose, use a pencil to mark your group number.
3. Your TA will carry out the transfer and you will observe the process. Please be careful to:
a. Wear gloves when you handle nitrocellulose.
b. Put the nitrocellulose on the anode side.
c. Remove all air bubbles between filter papers, nitrocellulose and gel.
Please make sure that you understand why.4. After transfer, your blot will be stored in dH2O at 4oC for immunoblotting.
April 5 Antibody incubation
Part 3. Western blot
Solution:
TBST (for 1 L):
NaCl 9 g
Tris base 6 g pH to 7.4
Tween 20 0.5 mlBlocking Solution:
1% gelatin in TBSTSubstrate Solution:
0.48 mM 4 chloro-1-naphthol
50 mM Tris-HCl
0.2 M NaCl
17% methanolPlease wear gloves when you handle nitrocellulose blot to avoid leaving your fingerprint on the blot.
1. Your blot has been stored in a baggie at 4oC since April 3. Rinse your blot with TBST twice. To do that, drain and discard the liquid in the baggie, and then add ~20 ml TBST. Seal the baggie, rotate it a few times, and discard the solution. Repeat one more time.
2. Blocking: Add 20 ml blocking solution to your blot. Please get rid of most of the air bubbles from the baggie before you seal it and make sure that the blocking solution covers the membrane. Place on a shaker for 40 min at room temperature.
3. Primary antibody blotting: The primary antibody is mouse anti-BSA antibody. Dilute the antibody 1:500 with the blocking solution to make up the antibody solution. To do this, add 20 ml of the antibody to 10 ml of the blocking solution. Discard the blocking solution, and add 10 ml of the antibody solution to your blot in the baggie. Get rid of all bubbles and seal the baggie. Rock the blot for two days at 4oC.
April 7 Development of the blot
4. Discard the antibody solution. Wash your blot with TBST 3 times, 3 min each. To do this, drain and discard the liquid in the baggie, and then add ~20 ml TBST. Seal the baggie, rotate it for 3 min, and discard the solution. Repeat two more times.
5. Secondary antibody blotting: The secondary antibody is HRP-conjugated goat anti-mouse IgG antibody. Dilute the antibody 1:1000 with TBST solution to make up the secondary antibody solution. To do this, add 10 ml of the antibody to 10 ml TBST solution. Add 10 ml of the antibody solution to your blot in the baggie. Get rid of all bubbles and seal the baggie. Place on the shaker for 40 min at room temperature.
6. Wash the blot with TBST 3 times, 3 min each.
7. Developing the blot: Prepare the substrate for HRP (4-chloro-1-naphthol) immediately before use. To do this, add 4 ml of 30% of H2O2 to 12 ml of the chloronaphthol solution. Mix the substrate solution and add it to your blot. Make sure the substrate solution evenly covers your blot. Let it develop for 1-5 min until the desired color is obtained. Purple bands will indicate immunoreactive proteins.
8. Photocopy your blot for your lab report.
Reading
Immunobiology A-18Study Questions:
1. What is the single major difference between the stacking gel and the separating gel, and what purposes do they serve?
2. When you run a SDS-PAGE gel or transfer proteins from a gel to nitrocellulose, in which direction do proteins move in an electrical field, from negative to positive or from positive to negative? Why?
3. This figure shows the distribution of molecular weight markers separated on a 10% gel. If the molecular weights of proteins that we are interested in are between 150 to 250 kDa., what percentage gel (higher or lower than 10%) should you use? Explain why.
4. Before blotting nitrocellulose with an antibody, we incubated the nitrocellulose with a blocking solution. The blocking solution contains gelatin, Tween 20, NaCl and Tris-HCl. Please describe the purpose each of the components serves.
5. List two ways in which ELISA is similar to Western blotting and two ways in which ELISA is different from Western blotting.