
How we make sense out of the whole group?
How do we decide what organisms to group together?
Can we find a way of categorizing and classifying mammals that sets them apart as a distinct taxonomic group and shows the evolutionary relationships among the different kinds of mammals?
Recognized that fossils represented organisms that were once alive.
| WORKSHEET: Historical definitions |
| "-idae" | indicates it's at the level of Family |
| "-inae" | indicates it's at the level of Subfamily |
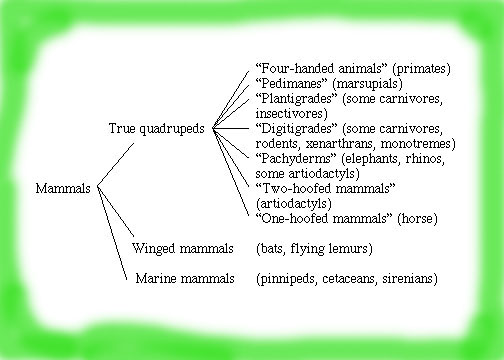
Think-Pair-Share: What other types of observable characteristics could you use to classify mammals? Are any of these categories any better or worse than any other types of observable characteristics?
taxonomy=the science of naming and classifying organisms
| Unguiculata | share many primitive features, evolutionarily ancient, diverse specializations | shrews, bats, primates, flying lemurs, anteaters, sloths, etc. |
| Glires | rodents, rabbits and hares | |
| Mutica | aquatic | whales and dolphins |
| Ferungulata | carnivores, aardvark, elephants, hyraxes, sirenians, perissodactyls (odd-toed ungulates) and artiodactyls (even-toed ungulates)
|
systematics= the scientific study of the kinds and diversity of organisms and of any and all relationships among them
phylogenetics=systematics, with the organizing principle being evolution

| WORKSHEET: Pruned trees |
Advantages
Disadvantages
Advantages
Disadvantages
Advantages
Disadvantages
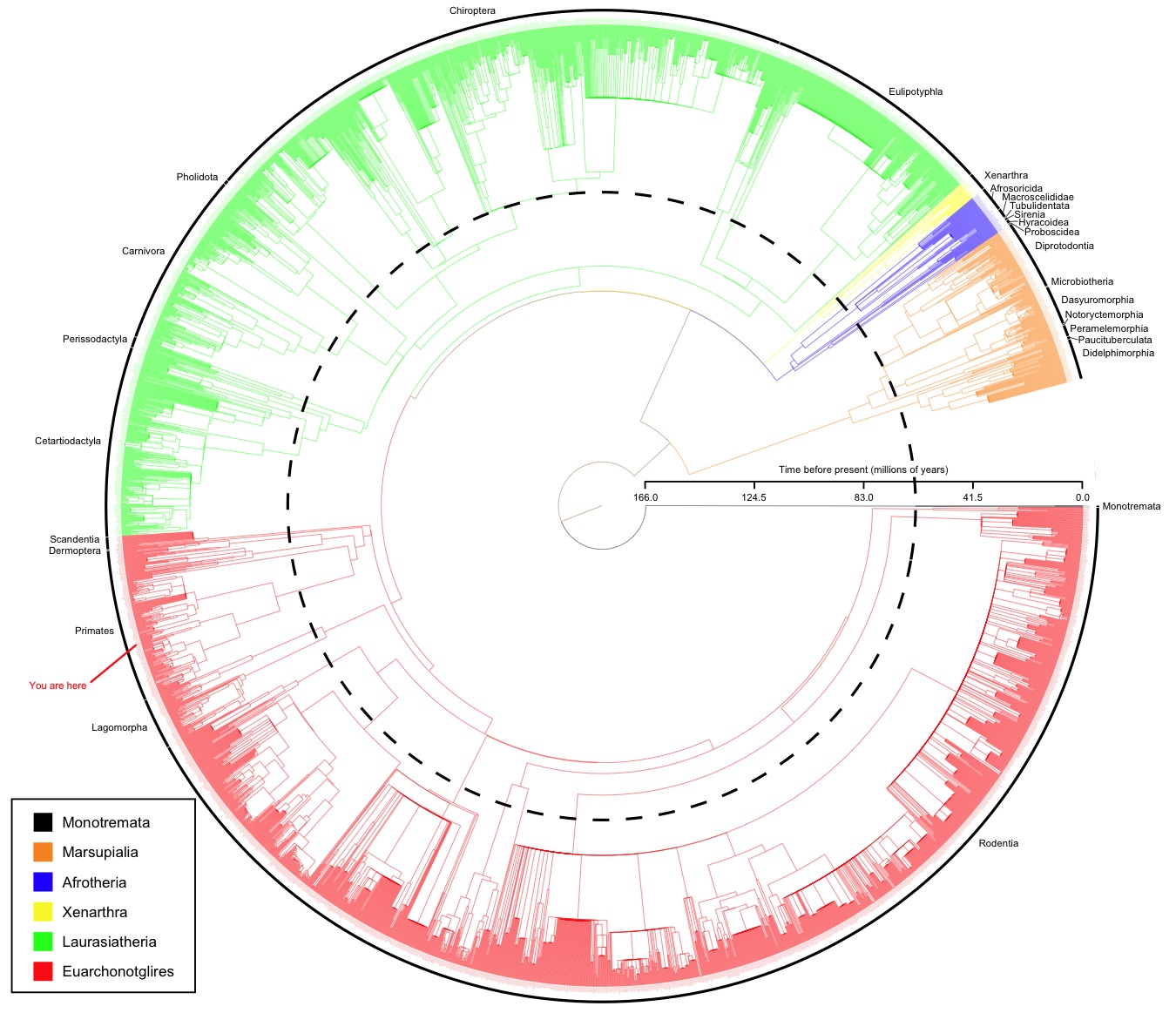
| WORKSHEET: Modern phylogenetic trees |

{kind=link}
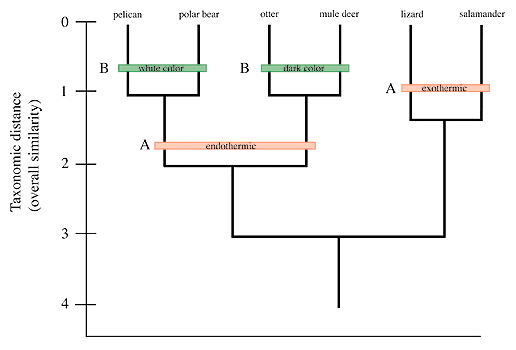{kind=link}
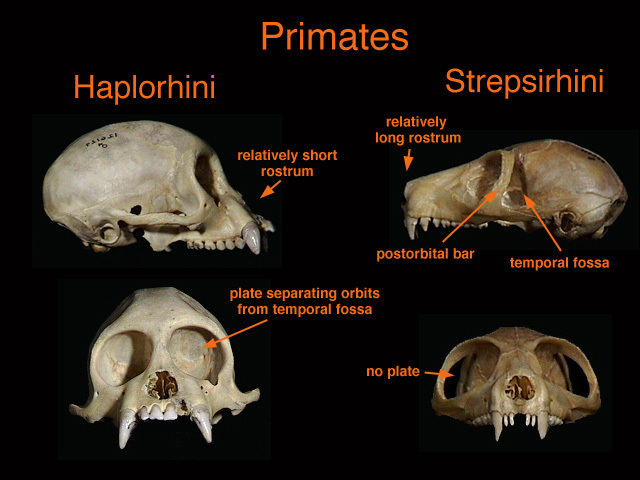{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}